
UNDERSTANDING HEREDITARY
ATTR-CM (hATTR-CM )
認識遺傳型 ATTR-CM (hATTR-CM )
WHAT IS hATTR-CM ?
- ~75%
-
About 75% of patients with hATTR amyloidosis exhibited cardiomyopathic features of the disease.3
- >30%
-
Carpal tunnel syndrome is also common and can often be the initial symptom in more than 30% of
hATTR-CM patients, with cardiomyopathy symptoms following as the disease progresses.6
AN OVERVIEW OF hATTR-CM
hATTR-CM 的概述

PATIENT CONSIDERATIONS
- African American, African, or
Afro-Caribbean descent (V122I)5,7,8 - Irish descent (T60A)8
- Men and women5
- Symptom onset may occur as early as 50-60 years of age9*
PROGNOSIS
Once diagnosed, untreated patients have a median survival of ~2 to 3 years.8
COMMON CHARACTERISTICS THAT MAY PRESENT IN hATTR-CM PATIENTS:
- Heart failure5
- Peripheral dysfunction (eg, peripheral sensorimotor dysfunction, peripheral neuropathy)5
- Autonomic dysfunction (eg, autonomic neuropathy, gastrointestinal complaints, unexplained weight loss, orthostatic hypotension, sexual impotence)5,9
- History of bilateral carpal tunnel syndrome5
*Age of onset varies depending on the causative mutation.
RECOGNITION OF CARDIAC INVOLVEMENT IN HEREDITARY AMYLOIDOSIS (VIA NT-proBNP MEASUREMENTS) IS CRITICAL, AS CARDIAC INVOLVEMENT IS ASSOCIATED WITH ADVERSE OUTCOMES AND WORSE OVERALL SURVIVAL, REGARDLESS OF PHENOTYPE.10
NT-proBNP, N-terminal pro-B-type natriuretic peptide.
SELECTED GENOTYPE-PHENOTYPE
ASSOCIATIONS IN hATTR AMYLOIDOSIS*
關於 hATTR 類澱粉沉著症之特定基因型與表現型的關聯性*
In hATTR amyloidosis, the clinical manifestation depends on mutation type, geographic origin of the patient, patient ethnicity, and other factors.11
Select each individual mutation type
for more information on clinical manifestations, age of onset, and patient heritage.
Manifestations
Age of Onset
Patient Heritage
Japanese 10
Manifestations
Age of Onset
Patient Heritage
Manifestations
Age of Onset
Patient Heritage
English13
Manifestations
Age of Onset
Patient Heritage
Manifestations
Age of Onset
Patient Heritage
Manifestations
Age of Onset
Patient Heritage
African8
Afro-Caribbean8
About 75% of patients with hATTR amyloidosis exhibited cardiomyopathic features of the disease.3
*This spectrum of genotype-phenotype associations is not comprehensive.
DIAGNOSTIC URGENCY
診斷的迫切性
In patients with
ADVANCED hATTR-CM IN UNTREATED PATIENTS IS ASSOCIATED WITH RAPID PROGRESSION, SERIOUS CARDIAC COMPLICATIONS, AND A WORSE MEDIAN SURVIVAL.5,8,19 THEREFORE, EARLY DETECTION IS CRITICAL, AS SURVIVAL WORSENS AS hATTR-CM ADVANCES.19
COULD HEART FAILURE IN YOUR AFRICAN AMERICAN PATIENTS BE A CAUSE OF hATTR-CM ?

In the United States, the most common mutation causing the cardiac form of the disease

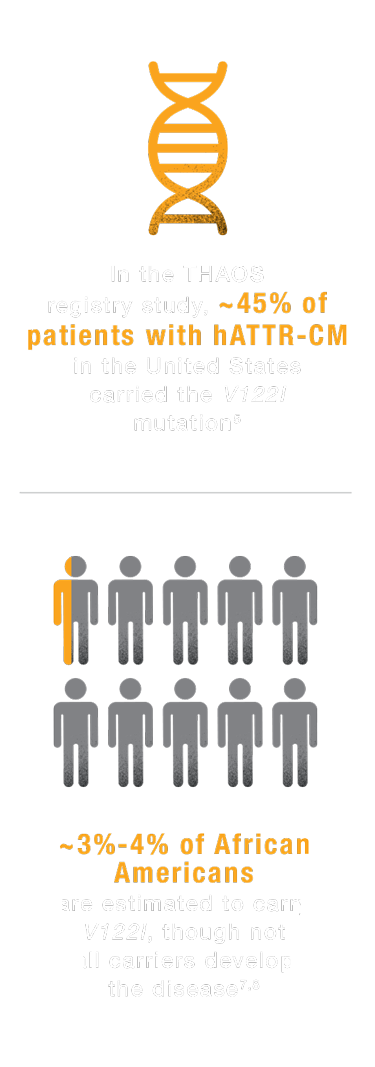
THAOS, Transthyretin Amyloid Outcome Survey.
V122I PRIMARILY PRESENTS WITH CARDIAC MANIFESTATIONS
In 1 study, the most common clinical presentation with

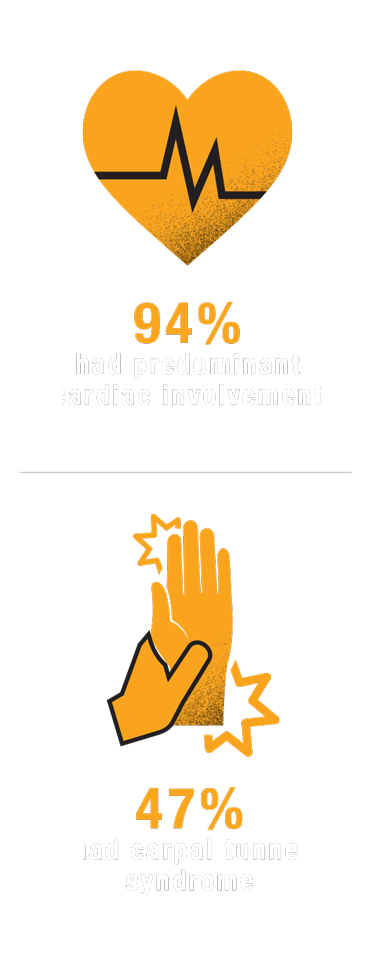
hATTR-CM IS OFTEN UNDERRECOGNIZED, AND INDIVIDUALS OF AFRICAN DESCENT WITH hATTR-CM DUE TO V122I CAN BE ESPECIALLY OVERLOOKED.20,22 IT IS IMPORTANT TO DISTINGUISH THESE PATIENTS FROM THOSE WITH OTHER FORMS OF CARDIOMYOPATHY AND HEART FAILURE.22
V122I PATIENT PROFILE
A CLINICAL PROFILE FOR A 67-YEAR-OLD MAN DISPLAYING THE CLINICAL SIGNS OF hATTR-CM

Family history
- Mother died due to complications from heart failure at age 63
- Brother suffered a stroke
Presentation and medical history
- Currently experiencing shortness of breath and fatigue
- History of hypertension, although blood pressure is controlled with appropriate medication
- Echocardiographic evidence of increased left ventricular wall thickness
- History of carpal tunnel syndrome with bilateral surgical repair
Diagnosis
- AL amyloidosis was ruled out
- Cardiac imaging using 99mTc-PYP was consistent with
ATTR-CM † - Genetic testing identified the V122I mutation of the TTR gene, consistent with the hereditary form of
ATTR-CM
*Hypothetical profile. Not an actual patient.
†99mTc-PYP is not FDA approved for the diagnosis of
99mTc-PYP, 99mtechnetium-labeled pyrophosphate.
JOE’S MEDICAL AND FAMILY HISTORY SHOULD RAISE SUSPICION OF hATTR-CM—A PROGRESSIVE, LIFE-THREATENING DISEASE22
UNDERSTANDING GENETIC RISK
了解遺傳風險
DIAGNOSIS OF hATTR-CM IN AN INDEX PATIENT SHOULD PROMPT GENETIC COUNSELING AND TESTING OF FAMILY MEMBERS15
- Early identification of
hATTR-CM is important to enable timely treatment15 - Genetic counseling and testing can be especially beneficial in at-risk family members, such as siblings or children of an index patient15
TIMING OF GENETIC TESTING15
- A targeted approach may enable diagnosis of disease upon the first detectable sign or symptom


*Percentage of individuals with a mutation who exhibit clinical symptoms.
LIFE-THREATENING, UNDERRECOGNIZED, AND UNDERDIAGNOSED, IT IS VITAL TO RECOGNIZE THE CLUE OF CLUES OF
References: 1. Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215-219. 2. Sekijima Y. Hereditary transthyretin amyloidosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, eds. GeneReviews [Internet]. Seattle WA: University of Washington; 1993-2019. 3. Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520-528. 4. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. 5. Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161-172. 6. Kapoor M, Rosser AM, Laura M, Reilly MM. Clinical presentation, diagnosis, and treatment of TTR amyloidosis. J Neuromuscl Dis. 2019;6(2):189-199. 7. Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22(3):171-174. 8. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017;135(14):1357-1377. 9. Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398-408. 10. Klaassen SHC, Tromp J, Nienhuis HLA, et al. Frequency of and prognostic significance of cardiac involvement at presentation in hereditary transthyretin-derived amyloidosis and the value of N-terminal pro-B-type natriuretic peptide. Am J Cardiol. 2018;121(1):107-112. 11. Nativi-Nicolau J, Maurer MS. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018;33(5):571-579. 12. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387-404. 13. Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28(1):10-21. 14. Rapezzi C, Longhi S, Milandri A. Cardiac involvement in hereditary-transthyretin related amyloidosis. Amyloid. 2012;19(suppl 1):16-21. 15. Conceição I, Damy T, Romero M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26(1):3-9. 16. Donnelly JP, Hanna M. Cardiac amyloidosis: An update on diagnosis and treatment. Cleve Clin J Med. 2017;84(12)(suppl 3):12-26. 17. Coelho T, Ericzon B, Falk R, et al. A Guide to Transthyretin Amyloidosis. Clarkston, MI: Amyloidosis Foundation; 2016. http://www.amyloidosis.org/wp-content/uploads/2017/05/2017-ATTR-guide.pdf. Accessed October 28, 2019. 18. Conceição I, Coelho T, Rapezzi C, et al. Assessment of patients with hereditary transthyretin amyloidosis – understanding the impact of management and disease progression. Amyloid. 2019;26(3):103-111. 19. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799-2806. 20. Damrauer SM, Chaudhary K, Cho JH, et al. Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of African or Hispanic/Latino ancestry. JAMA. 2019;322(22):2191-2202. doi:10.1001/jama.2019.17935 21. Connors LH, Prokaeva T, Lim A, et al. Cardiac amyloidosis in African Americans: Comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158(4):607-614. 22. Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709-716. 23. Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-122. 24. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-2594.